Qu1: D
Question about geometry
goes back to hybridisation
and geometries of
some N systems. The only difference between N and C systems is that you
have to remember to count the lone pair on N as it requires an orbital
and therefore space - count it as a group when deducing the
hybridisation. i is an amine, N = sp3, bond angles
of about 109.5o,
ii
is a nitrile, N = sp, bond angle is 180o, and iii
is an imine, N = sp2, bond angles of about 120o, therefore
giving : ii > iii >
i
Qu2: AB
Relates to hybridisation
and nomenclature.
i is an alkane, C
= sp3, therefore 25% s character, ii is an alkene
C = sp2, therefore 33% s character and iii is an alkyne,
C = sp, therefore 50% s character. Increasing the s character in the
hybrid orbital makes it smaller and creates a stronger interaction with the
H atom and so a stronger bond. Therefore iii > ii >
i
Qu3: E
Acidity
.... Know your pKa's or work it out ? If you need to work
it out, then consider the general acidity equation HA <=>
H+
A-. Look at the factors that stabilise the conjugate base,
A-. First notice that we have two C-H systems and an S-H
system.
i is a ketone (pKa = 19) where
the conjugate base has the negative charge on a C atom but resonance
can further stablise it to an electronegative O atom - remember
spreading charge out (delocalisation) is a
stabilising effect. ii
is an alkene (pKa = 40), this time the conjugate base will have
the negative charge on the C atom - but the charge can not be
delocalised so there is no further resonance stabilisation.
Finally iii , a thiol (pKa =
10) - the conjugate base puts negative charge on the S atom, S is in
the same group of the periodic table as O, but one row lower. Remember
that acidity increases as you go down a group since the larger atom can
readily accommodate the extra charge. So overall we have iii
> i > ii
Qu4: B
Calculate the formal
charges for the O atom = group number - number
of bonds - lone pairs electrons or by relating to known structures : i
= +1 based on 6 - 3 - 2,
ii
= -1 based on 6 - 1 - 6 and iii = 0 based on 6 - 2 - 4.
Therefore i > iii > ii
Qu5: B
Physical
properties relate to intermolecular forces... for alkanes this
amounts to weak induced dipole interactions. The shape of the molecule
affects the surface areas that are in contact. More branched structures
are more spherical in shape and therefore have less surface area in
contact. Larger surfaces in contact means greater interaction and hence
a higher boiling point. There is NO relation between thermodynamic
stability and physical properties. So i > iii
> ii
Qu6: A
Remember the rules
for ranking resonance structures : complete octets are most
important. Here we focus on the C=C-O unit. Both i and ii have
complete octets at both C atoms and O (despite the charges in ii) and both have the same number of
bonds. iii has an incomplete
octet on C and has one less bond than the others. So i > ii
> iii
Qu7: B
Acidity.... which functional group is more acidic ? Why is this
important ? The more acidic H will be removed first when reacted with a
base. Here we have a phenol (pKa = 10) and a carboxylic acid (pKa = 5),
so the H in the acid group will be removed first. Since only 1
equivalent of base has been added, i will
be the major species in solution. Some of iii will also be formed but ii is impossible since the more
acidic COOH group will protonate the adjacent phenoxide O- : i
> iii
>
ii
Qu8: A
Oxidation
states...count the bonds attached to the C,
each H counts +1, C counts 0 and a bond to a more electronegative atom
(e.g. O, N, X) counts -1.
Total the count and then since the atoms
are neutral, just switch the sign since the oxidation state for the C plus the groups attached must
equal 0. In i the C is
attached to 4 Cl (count - 4) therefore oxidation state C = +4. In ii the C is attached to H (count
+1), two bonds to O (count -1 per bond) and C (count +0) therefore
oxidation state C = +2.
In iii the C has two bonds to
C (count 0 per bond), a bond to H (count +1) and a bond to O (count -1
per bond) therefore oxidation
state C = 0.
Therefore i > ii > iii
Qu9: A
Basicity...
the strongest base will create the most of the
conjugate base. Compare to O systems first... O- is a stronger base
than O, so ii > iii. Now compare N- and O-
. N is less electronegative than O so it is a better electron
donor and hence a better base (Lewis definition) so i > ii. Therefore overall i
> ii > iii
Qu10: D
Resonance
stabilisation... look at how the -ve charge can be
delocalised. In all 3 cases it can be delocalised into the ketone group
on the left. In i this is the
only thing that happens. In ii and
iii it can be delocalised due
to the extra pi systems present, in ii
the -ve charge can be delocalised to an electronegative O atom
(good !) and in iii to
a C atom (not as good), so overall ii >
iii > i
Qu11: B
From the extraction experiment. Seems a hard question needing complex
math,
but if you understand the principles, it's really pretty straight
forward. Given
that KD = 2 chloroform : water and the solvent volumes are
in the ratio 1 : 1
so there will be half as much in the water layer as the extracting
dichloromethane layer i.e. 1
: 2 ratio. This means that each
dichloromethane extraction will extract 2/3 of the total. So the
first extraction takes out 67% than the second takes 2/3 of the
remaining 33% = 22%, therefore in total 67 + 22 = 89 % will have been
extracted.
Qu12: E
From the physical properties experiment. Remember boiling points
increase
with increasing pressure so they are higher at sea level than here in
Calgary
at almost 4000ft above sea level. Rough
rule of thumb for Calgary is 1o
for every 15o above 50o. Thus for 200o
we are talking about a 10o increase.
Qu13: C
A yield calculation. Reaction stoichiometry 1:1. 1.38g of
salicylic acid (138 g/mol) is 10 mmoles. 1.0g of acetic anhydride
(102 g/mol) is 9.8 mmoles, so the anhydride is limiting. 1.2g of
aspirin (180 g/mol) or 6.67 mmoles was obtained, so the yield is 6.67 /
9.80 = 68%.
Qu14: D
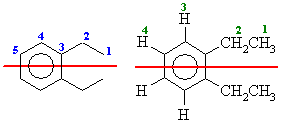
From the molecular models experiment. First draw out 1,2-diethylbenzene
(if you can't, then you need to work on your nomenclature).
The number of types of C are shown below in blue on the left and the H are shown
below in green on the right. The red line shows the symmetry due to the mirror
plane that bisects the molecule.
Qu15: AB
From the molecular models experiment on IHD.
Either use the molecular structures and count pi + r or use the
equation based on the molecular formula (see link). Benzene has 3 pi
bonds and one ring so it has an IHD = 4. Cubane is more
difficult, but the IHD for C8H8 is 5.
Qu17: C
Oxidation
states
C2 has the following bonds : 1 x C (count 0) and 3 x O (count
-3) so the
sum = -3 therefore the oxidation state of C2 = +3.
C13 has the following bonds : 1 x N (count -1), 1 x C (count 0)
and 2 x H (count +2) so the
sum = +1 therefore the oxidation state of C13 = -1.
Qu18: D
The functional
group in question is C-O-C so it's an ether.
Qu19: D
The functional
group in question is -C(=O)-O-C so it's an ester.
Qu20: D
IHD....Unsaturation
arises due to pi bonds or rings. So count these up
in the structure: there are 6 C=C and 2 C=O plus 3 rings (2
x 6 membered and 1 x 7 membered), so 11 in total.
Qu21: E
O12 is part of a double bond,
C=O, so it is sp2. N10
is sp2 because the lone pair on the N is involved in resonance with the
adjacent pi system, a C=O, it's an amide N. N15 is sp3 because it has 3 C atoms
attached, plus the N lone pair but there is not resonance since there
is not adjacent pi system, it's an amine N.
Qu22: B
Hydrocarbon
hybridisations.... C2 is
part of a double bond,
C=O, so it is sp2. C5 is
sp3 because it has 2 x C, 1 x O and 1 x H atoms attached. C17
is sp3 because it has 3 x H and 1 x O atoms attached.
Qu23: B
Apply the Cahn-Ingold-Prelog
rules at the 2 chirality centers. C5 priority order : -OC >
CS > C(=O) > H, the low priority H is already at the back, 1-2-3
order goes clockwise => R. C6
priority order : -S > CO > CC > H, the low priority H is
currently at the front, so take care. When H is at the back, the 1-2-3
order goes counter clockwise => S.
Qu24: D
Basicity
by the Lewis definition is lone pair donation, so can look
at the availability of the lone pairs. Neutral C atoms with 4 sigma
bonds don't have lone pairs so they are not basic. N is less
electronegative than O so N is a better electron donor and hence a
better base than O. N10
is in an amide and N15 in an
amine. The lone pair in the amide is also involved in resonance with
the C=O so making it less available to react as a base. So the amine N,
N15 is the most basic site.
Qu25: B
The conformation shown is an eclipsed
conformation since the bonds on adjacent atoms are aligned. Here the
two principle groups, the methyl groups are at 120o
torsional angle. This conformation does not have a more specific
name. Anti means
a 180o torsional angle and syn a 0o torsional
angle.
Qu26: C
The conformation shown is a staggered
conformation since the
bonds on adjacent atoms have torsional
angles of 60o.
Here the two principle groups, the methyl groups are at 180o
torsional angle, so this is an anti
conformation. A syn
conformation has a 0o torsional
angle and gauche
has them at 60o. Trans is used to
described substituents on cyclic structures or across double bonds.
Qu27: AE
The two terms that apply to
cyclohexane locations are axial
and equatorial. Substituents
prefer to be equatorial to reduce the steric strain due to Van
der
Waals strain due to 1,3-diaxial
interactions and torsional
strain that
occur when they are axial. The chair
is the more stable conformation (staggered therefore less torsional
strain) compared to the boat, so B and
D must be incorrect. A ring
flip of A will have two
axial methyl group so A is the
more stable conformation. C
ring flips to
give two equatorial methyl groups which will be lower energy and hence
more stable. E ring
flips to give two axial and one equatorial methyl groups which will be
higher energy and hence less stable than the conformation shown.
Qu28: CE
First draw out each structure or work out the molecular formula of each,
if you can't do this, you should review your nomenclature.
If they have the same molecular formula, then they are isomers. 2-butanone
is a ketone = C4H8O and so has 1 unit of unsaturation.
2-butanol is a saturated alcohol = C4H10O. Diethyl ether
is saturated, C4H10O. Butanal is an aldehyde = C4H8O
and so has 1 unit of unsaturation. Butanoic acid is a carboxylic acid
= C4H8O2. Cyclopropylmethanol has a cyclic
unit and is an alcohol = C4H8O.
Qu31: D
Torsional
strain is due to the alignment of bonds on adjacent atoms. A is angle strain, B and C are Van
der
Waals strain and E is ring
strain.
Qu32: C
The chair
is the most stable and the boat
is the least stable of these conformations with the twist boat in
between. The chair is all staggered whereas the boat is partially
eclipsed. The twist
boat is more stable than the boat because the slight twist relieves
a little of the torsional
strain.
Qu33: C
Longest chain is C10, contains a C-C only so a decane. This means
it has to be C or E. E
is wrong because the methyl group in the complex branch is on C2 and
not C1 as the name implies.
Qu34: C
A ketone group at C1 (priority functional group), then use first point
of difference to get the
methyl group at C3, ethyl at C5. Remember to alphabetise ignoring the
"di" so ethyl before dimethyl.
Qu35: C
The compound is an ester, -C(=O)-O not an ether - the carbonyl
group makes a difference and the carbon of the carbonyl is C1. So
it can not be E. The acid
part of the ester (left of C=O as drawn) has 5C and contains a C=C
starting at C2 so it's a pentenoate.... that rules out A and B. The alcohol part of the ester
(right of the C=O as drawn) is from isobutanol. The double bond has E
stereochemistry. Hence isobutyl (E)-3-methyl-2-pentenoate.
Qu36: E
Alkene stereochemistry as described by E and Z. The longest chain
including the two C=C is 4 carbons so we need a butene. This limits
our choice to C, D or
E. The double bond has E stereochemistry because the higher
priority groups (the OCH3 and the CH3) are on the opposite sides of the
double bond. Use first point
of difference to get the chloro group at C. Hence we have
(E)-1-chloro-2-methoxybutene = E.
Qu37: E
Meta implies we have a pair of 1,3-substituents so that
restricts us to B, D
or E. Nitro = -NO2 and phenol implies a
-OH substituent on the benzene ring. A is ortho-nitrophenol, B does not have a nitro group, NO
is a nitroso. C does not
have a nitro group or an -OH, D
is meta-aminophenol.
Qu38: C
Use the descriptors trans- and (1,2) and look at the position
of the two methyl groups.... A trans-(1,4)-, B trans-(1,3)-,
C
trans-(1,2)-,
D cis-(1,2)-, and E is cis-(1,3).
Qu39: C
-2-one ending implies we need a ketone. This narrows the choices to A, C
or D. B and E are aldehydes. The name is a
pentenone so we need a C5 chain including the C=O and a C=C. D is wrong because it's a C6
chain.
In terms of assigning
configurations, the group order is -OH > C=O > C=C >
H. Remember to assign the sense of the rotation to the order of
the groups when the H is away from you.... A is R, we need S.
Qu40: B
Did you look at the nomenclature
of bicyclics ? Bicyclic means two fused rings. The [3.2.1] means that there
are 3C, 2C and 1C in the links between the shared (bridgehead) C atoms. This
gets rid of C, D
and E which are [2.2.1], [2.2.2] and [2.2.2] respectively. Then we number
from one of the shared C bridgehead atoms via the longest link first so as to
give the hydroxyl group (the first point of difference) the lowest number.
![[Chem 350 Home]](../mol.gif)