Chem 351 Final Fall 2023
Here is an post-mortem analysis / "how to" for
the FINAL. The questions are split by the sections. At the start of each section
are a few suggestions of what to look for or how to tackle the question type.
RELATIVE PROPERTIES:
Identify the controlling feature,
which is not always as obvious as
it may appear. Look for two pairs of similar systems to compare that
have
minimal differences in structure.
| Qu 1: |
Stability of the carbocation : we have a tertiary benzylic carbocation (resonance stabilised), an aryl cation and a primary cation. In stability order they are tertiary benzylic > primary > aryl. |
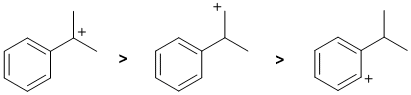 |
| Qu 2: |
Basicity... The stronger the base, the more deprotonation of the terminal alkyne there will be.
Let's first compare the two O systems: in one, the O is next to a pi system... so the lone pairs will be involved in resonance with the pi system making them less available and thus less basic than in the other cyclohexyl system. Now compare the -ve N and the -ve O substituted cyclohexanes (where the -ve atoms are in the same row of the periodic table). In simple systems, N is more basic than O because it is less electronegative (e.g. consider the pKas for ROH = 15 and RNH2 = 35 where R are alkyl groups, ArOH = 10). |
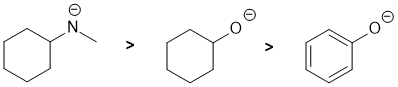 |
| Qu 3: |
Nucleophilicity... let's first compare the S to the O system where the -ve atoms are in the same group of the periodic table... O being smaller makes it less nucleophilic than S (recall that nucleophilicity increases as you go down a group). Now compare the N and the O where the atoms are in the same row of the periodic table. Note that we have -ve O and neutral N so the -ve O is the stronger nucleophile. |
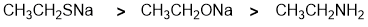 |
Qu 4: |
In terms of the reaction, we are looking at elimination of alcohols with H2SO4 / heat... These are typically E1 and the rate is dictated by the stability of the carbocation being formed. Drawing out these intermediates we will get a secondary benzylic carbocation (resonance stabilised), an aryl cation and a primary cation. In stability order they are secondary benzylic > primary > aryl which dictates the relative reactivity. |
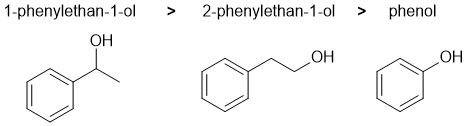 |
Qu 5: |
Using the C NMR tables, ketones C=O should be 190-220ppm, ester C=O should be about 155-190ppm and sp3C-O would be 40-85ppm. |
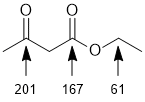 |
Qu 6: |
The series of C-H bonds differ due to the changing hybridisation of the C atom. Typical values are sp C-H 3300 cm-1, sp2 C-H 3100 cm-1 and sp3 C-H 2900 cm-1 which aligns with stronger bonds having higher frequencies (think of the Hooke's Law model) (see IR tables) |
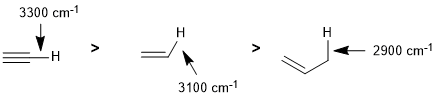 |
Qu 7: |
Counting types of carbon |
 |
| Qu 8: |
The reaction here is dehydrohalogenation of an alkyl bromide using a strong base (NaOEt) and heat. These reactions are E2. In terms of Zaitsev's rule, increasing the steric hindrance in the alkyl bromide at the removed H atom will decrease the more highly substituted alkene (more stable alkene, Zaitsev product) so giving more of the anti-Zaitsev product. |
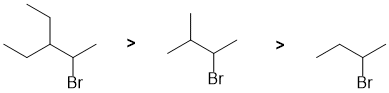 |
MOLECULAR PROPERTIES
No real method here, really just do you know various aspects of molecular structure / reactions, applied to each of the questions.
| Qu9: |
The reaction here is radical substitution (an alkane reaction) of an alkyl group sp3 C-H to a C-Br. The typical reagents for this reaction are Br2 and heat or uv light. |
|
| Qu10: |
Cahn-Ingold-prelog rules are based on the heavier atom at the first point of difference. In terms of alkene geometry, in terms of priorities, at the left end we have Cl >C and C>H at the right end. Given that the higher priority groups (i.e. the Cl and the C) are on the same side (they are together), this is a Z isomer. |
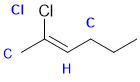 |
| Qu11: |
The reaction here is a Williamson ether synthesis which is an example
of a nucleophilic substitution (SN2) and they are often in competition with elimination (i.e. nucleophilicity vs basicity). Since a methyl bromide can't undergo a 1,2-elimination and tertiary bromides aren't great for SN2, the reaction of t-butoxide with methyl bromide is the better choice. |
|
| Qu12: |
The H-NMR chemical shifts of sp C-H and sp2 C-H are controlled by magnetic anisotropy caused by the pi systems. The sp2 C-H of an alkene is typically 4.5-7 ppm and sp C-H of an alkyne 2-3 ppm because the alkene H are deshielded due to the magnetic anisotropy. |
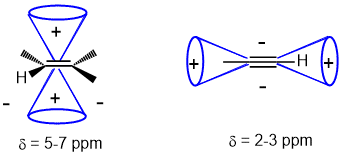 |
| Qu13: |
The of secondary alcohol will undergo an SN2 with thionyl chloride to give the alkyl chloride with inversion of configuration due to the backside attack (180 degree approach of the nucleophile). But this chloride then undergoes another SN2 with the bromide and inverts again, so back to the original stereochemistry. |
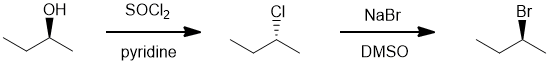 |
| Qu14: |
Resonance contributors can be derived by arrow pushing... |
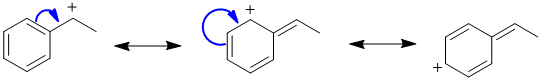 |
REACTIONS:
Three types of questions.
For those with starting materials work from the starting materials towards the products using the reagents to "see" what product to look for.
For those with the product work backwards... looking at the functional groups in the products to think about how you may have got there.
For those wanting reagents look at the functional groups in the starting material and products to try to determine what may have happened. Look at the reagents in each option to see what effect they would have on the SM...
| Qu15: |
We should work forwards... the product is an allylic bromide i.e. an alkene with an adjacent bromide. This suggests an elimination of the alcohol (i.e. strong acid/heat) then radical substitution of the allylic system (e.g. NBS / heat). |
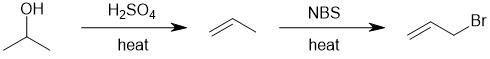
|
| Qu16: |
We should work forwards... the first reaction uses NaOEt which is a good base... and since we have a carboxylic acid, this is going to deprotonate to form the carboxylate. The carboxylate can function as a nucleophile in an intramolecular nucleophilic substitution (SN2) reaction with the primary bromide to form a cyclic ester. Count C atoms ! |
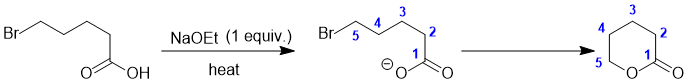
|
| Qu17: |
We should work backwards... step 2 is heating with NaOEt suggests an elimination of an alkyl halide to give the alkene in the product. Now step 1 is the conversion of an alcohol to an alkyl chloride via a nucleophilic substitution. The Cl on the benzene ring needs to be there from the beginning. |
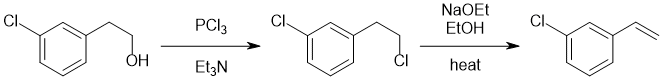
|
| Qu18: |
We should work forwards... the transformation is alcohol to alkene but we have the less highly substituted alkene (i.e. anti-Zaitsev ). Simple dehydration of the alcohol using acid/heat follows Zaitsev's rule and thus the wrong regiochemistry. This indicates that we need to convert the alcohol to an alkyl halide or tosylate and then heat with a bulky base... |
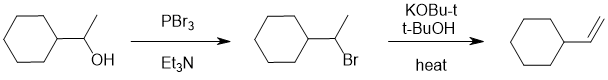
|
| Qu19: |
We should work forwards...the reaction is an SN1 involving the conversion of the alcohol to a rearranged alcohol via a nucleophilic substitution via a carbocation (i.e. SN1) that rearranges from secondary to tertiary (via a 1,2-hydride shift). Treating the alcohol with aq acid will allow the alcohol to protonate to make the better LG, generate the carbocation that will rearrange and then water to act as the nucleophile to reform the alcohol. |
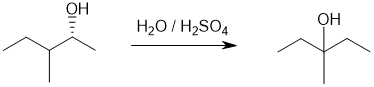
|
| Qu20: |
We should work backwards, we are doing making an ether via a Williamson ether synthesis. Since the Williamson process doesn't change the configuration at the alcohol center (since we don't make or break bonds to that C atom), we need to start with the two O groups trans and with the same configurations. |
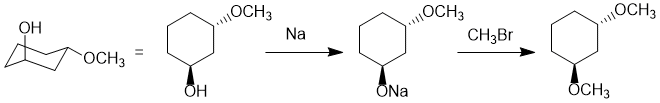
|
CONFORMATIONAL ANALYSIS:
Understanding the terminology and the
energies involved in
conformational
analysis.
Qu 21:
See the image for the 7 equatorial H atoms.
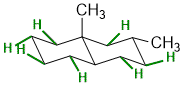
Qu 22:
Interpreting Newman projections... draw out the named structure first and see that it is a C6 structure. If one checks the C count in the Newman projections, one of them is C5. Then check the connectivity of the other structures looking for the connections of the 4 methyl groups...
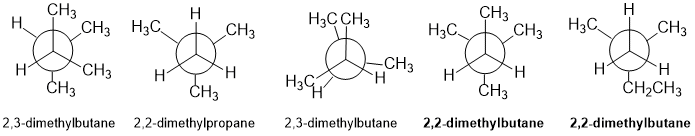
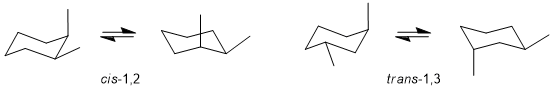
Qu 23:
Check which options are 1,3- and then which are also trans (i.e. the two substituents are on opposite sides). 1,3-trans will require one axial and one equatorial. The lowest energy conformation will have the larger substituent equatorial.
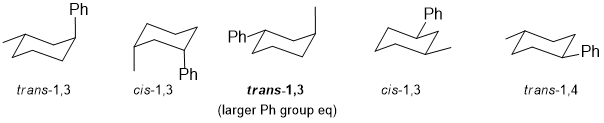
Qu24:
The two Cl atoms are at 120 degrees (torsional angle) in this eclipsed conformation.
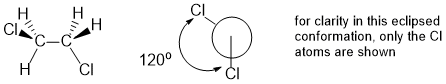
Qu25:
For the conformation to have the least strain it will be the most stable conformation so we need a staggered conformation with the methyl groups as far apart as possible (i.e. anti or 180 degrees).
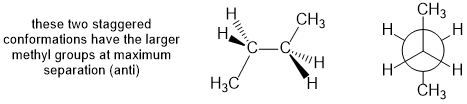
Qu26:
In order for the chair conformations to be of equal energy (i.e. like cyclohexane itself) and if substituted then there will need to be two groups that (1) need to be the same and (2) need to be one equatorial and one axial as in cis-1,2-dimethylcyclohexane or trans-1,3-dimethylcyclohexane.
Qu27:
The two structures are isomers (same MF) and have the same function group and same skeleton. Of the terms available, regioisomers is the best term (e.g. consider the alkenes as the elimination products from 2-pentanol).
Qu28:
In an amide, the N is sp2 hybridised due to the resonance interaction of the N lone pair with the C=O group so it is trigonal planar and hence the C-N-C bond angles will be about 120 degrees.
SPECTROSCOPY:
Use any IR information to get the
functional groups. Use the H-NMR
to get the number of types of H, how many of each type from the
integral
and what they are next to from the coupling patterns. Chemical shifts
should
tell you if the group is near -O- or maybe C=O groups etc.
| Qu29: |
IR shows a carbonyl (i.e. C=O) at 1718 cm-1 which is typical for a ketone as is the C-NMR peak at 209ppm. The H-NMR has 3 types of H. The 3H singlet at 2.1 ppm, suggests -CH3 group, and the peaks at 3H triplet at 1.1 ppm and 2H quartet at 2.5ppm together indicate a -CH2CH3 group. This pieces of data are only consistent with 2-butanone. |
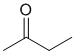
|
| Qu30: |
IR shows no carbonyl (i.e. C=O) near 1715 cm-1 or in the C-NMR (near 200ppm). The H-NMR has just 2 types of H. The peaks at 3H triplet at 1.0 ppm and 2H quartet at 2.4ppm together indicate a -CH2CH3 group. The absence of the carbonyl and only ethyl groups are consistent with the tertiary amine. |
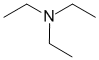 |
| Qu31: |
IR shows no carbonyl (i.e. C=O) near 1715 cm-1 or in the C-NMR (near 200ppm). The H-NMR has 3 types of H. The 3H singlet at 0.8 ppm, suggests -CH3 group, and the peaks at 3H triplet at 0.7 ppm and 2H quartet at 1.2ppm together indicate a -CH2CH3 group. Overall, no carbonyl and H-NMR shifts are more shielded are more likely to reflect a simple alkane. |
 |
| Qu32: |
IR shows a carbonyl (i.e. C=O) at 1792 cm-1 which is very high for a typical ketone. The C-NMR peak at 175ppm supports the carbonyl from a carboxylic acid derivative. The H-NMR has just 2 types of H. The peaks at 3H triplet at 1.2 ppm and 2H quartet at 2.9ppm together indicate a -CH2CH3 group. The high, single IR carbonyl suggest the acyl chloride. |
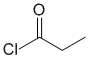 |
| Qu33: |
IR shows an -OH at 3400 cm-1 (broad) and consistent with the exchangeable singlet (11.7 ppm 1H) in the H-NMR. In addition, the IR shows a carbonyl (i.e. C=O) at 1716 cm-1. This information suggests a carboxylic acid. H-NMR shows 2 other types of H and the coupling and the integrals suggests a CH3CH2- group. |
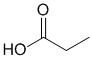 |
| Qu34: |
IR shows an -OH at 3300 cm-1 and consistent with the exchangeable 1H singlet (2.6 ppm) in the H-NMR. H-NMR shows 2 other types of H and the coupling and the integrals suggests a CH3CH2- group. |
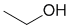 |