Chem 351 Final Fall 2023
Here is an post-mortem analysis / "how to" for
the FINAL. The questions are split by the sections. At the start of each section
are a few suggestions of what to look for or how to tackle the question type.
RELATIVE PROPERTIES:
Identify the controlling feature,
which is not always as obvious as
it may appear. Look for two pairs of similar systems to compare that
have
minimal differences in structure.
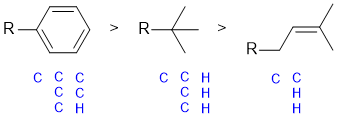
| Qu 1: |
Cahn-Ingold-prelog rules are based on the heavier atom at the first point of difference. In each case the point of attachment is a C atom, so we need to look at what atoms those C atoms are attached to. A good approach is to make a list of the attachments, listing them out in order of decreasing atomic number, then locating the first difference.
|
 |
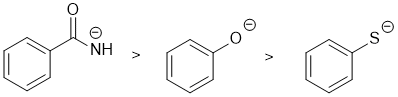
| Qu 2: |
Basicity... let's first compare the S to the O system where the -ve atoms are in the same group of the periodic table... O being smaller makes it more basic than S (recall that in terms of acidity RSH > ROH). Now compare the N and the O where the -ve atoms are in the same row of the periodic table. In simple systems, N is more basic than O because it is less electronegative (e.g. consider the pKas for ROH = 15 and RNH2 = 35 where R are alkyl groups, ArOH = 10, Ar-NH2 = 27). Here the N is also involved in resonance with the adjacent C=O pi system which makes it less basic (due to resonance stabilisation of the lone pair) but that doesn't overcome the electronegativity of O vs N (e.g. RNH2 = 35 vs RC(=O)NH2 = 15). |
 |
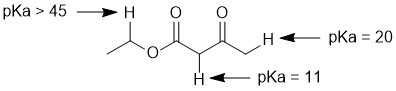
| Qu 3: |
Acidity.... Need to look at the stability of the conjugate bases and consider the factors that affect that stability. We have 3 CH systems that are adjacent to different groups. We have a methylene (CH2) between two C=O, a methylene next to an O and a methyl group (CH3) next to a C=O. A -ve charge on a C atom adjacent to a C=O will be stabilised by resonance (enolates) and two adjacent C=O will provide more resonance stabilisation that an single C=O. |
 |
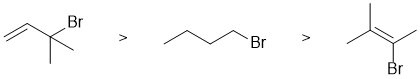
Qu 4: |
In terms of the reaction, we are told it is nucleophilic substitution... since we have a weak nucleophile (water), a good leaving group (Br) in a polar medium (water), this is likely SN1 and hence, it this question, we need to focus on the stability of the carbocation being formed. Drawing out the 3 bromides we will get a tertiary carbocation (also resonance stabilised), a vinyl cation and a primary cation. In stability order they are tertiary > primary > vinyl which dictates the relative reactivity. |
 |
Qu 5: |
Counting types of carbon |
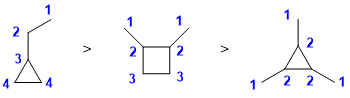 |
Qu 6: |
The number of lines = multiplicity (i.e. coupling) of the signals for each of the positions indicated is determined by the number of neighbours (n) that are of a different type and the n+1 rule. Here we see a singlet (0 neighbours), a triplet (1 neighbours) and a quartet (3 neighbours). |
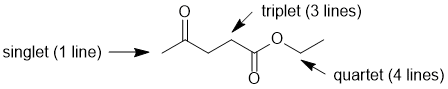 |
Qu 7: |
Since CC sigma bonds are stronger than CC pi bonds, the cycloalkane is more stable than the two alkenes. One factor in alkene stability is that a more substituted alkene is more stable, so the 3-hexene (disubstituted) is more stable than 1-hexene (monosubstituted). (PS : did you learn from your mistakes on the MT ? see Chem351F23 MT thermodynamics question) |
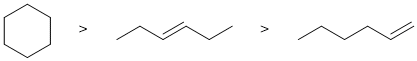 |
| Qu 8: |
Ranking resonance structures.. (1) either (a) maximise the number of atoms that satisfy the octet rule or (b) maximise the number of bonds (2) if there is charge separation is it in agreement with electronegativity. |
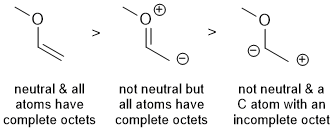 |
MOLECULAR PROPERTIES
No real method here, really just do you know various aspects of molecular structure / reactions, applied to each of the questions.
| Qu9: |
The C atom for the electrophile in simple nucleophilic substitution needs to be sp3 hybridised...so the benzyl chloride system make a good substrate for both SN1 and SN2 reactions (stable carbocation, but primary) |
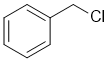 |
| Qu10: |
The of 1-bromopropane with a an alcohol in the presence of Na2CO3 is a nucleophilic substitution where the alcohol will be the nucleophile. Larger groups (sterics) tend to make nucleophiles less reactive, so ethanol will be more reactive than t-butanol. |
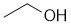 |
| Qu11: |
The reaction here is dehydration of an alcohol using a strong acid (the alkene products are shown). These reactions are E1 and follow Zaitsev's rule giving the more highly substituted alkene (more stable alkene) as the major product. |
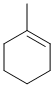 |
| Qu12: |
This relates to solubility and acid/base chemistry. The phenol (the protonated form) is non-polar, and is only a weak acid (pKa = 10) and it is not deprotonated effectively by the bicarbonate (solubility experiment). Since the phenol is non-polar (neutral) it will be dissolved in the ether layer. |
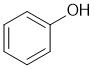 |
| Qu13: |
This relates to the chromatography experiment. More polar materials run more slowly on a normal phase TLC plate (because the plate is polar). The acetaminophen with more electronegative atoms and polar groups capable of H-bonding with the stationary phase will run more slowly and have a lower Rf. |
 |
| Qu14: |
This relates to the extraction experiment. If the KD = 3 and equal volumes are being used, then it will partition in a 3:1 ratio such that 3 = [ether]/[water] = 75% / 25% indicating 75% : 25% ratio so that 75% will be extracted. |
|
REACTIONS:
Three types of questions.
For those with starting materials work from the starting materials towards the products using the reagents to "see" what product to look for.
For those with the product work backwards... looking at the functional groups in the products to think about how you may have got there.
For those wanting reagents look at the functional groups in the starting material and products to try to determine what may have happened. Look at the reagents in each option to see what effect they would have on the SM...
| Qu15: |
We should work backwards... the product is an alkene and it's being created by heating with base... this suggests an elimination of an alkyl halide, in this case an alkyl bromide. The bromide has been formed by a radical substitution of a benzylic system, a simple hydrocarbon... count C atoms. |
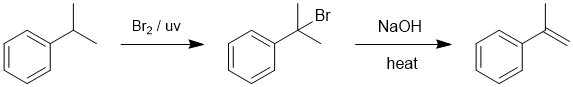 |
| Qu16: |
We should work forwards... the first reaction the conversion of the alcohol to the tosylate using tosyl chloride and then a nucleophilic substitution (SN2) of the tosylate with ammonia as the nucleophile to make the amine. The SN2 will cause an inversion of stereochemistry, so we need the amine with the opposite configuration to that at the -OH center in the starting material. |
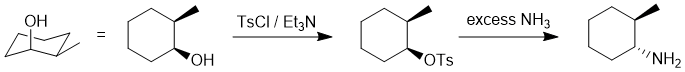 |
| Qu17: |
We should work forwards... the first reaction is an SN2 involving the conversion of the alcohol to the alkyl halide using phosphorous bromide via a nucleophilic substitution with inversion of stereochemistry followed by a second SN2 of the alkyl bromide to the thiol with a second inversion (so back to the original stereochemistry). |
 |
| Qu18: |
We should work forwards...but starting with a review of the overall transformation by comparing starting material and product. Both the starting material and the product are aldehydes but the product has gained +1C. This suggests the formation and then alkylation of an enolate. This would require a strong enough base (aldehyde enolate pKa = 17) and then likely a methyl halide. |
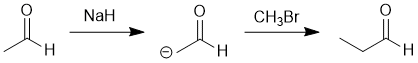 |
| Qu19: |
We should work forwards... the reaction is an SN1 involving the conversion of the alcohol to the alkyl halide using HCl via a nucleophilic substitution via a carbocation that rearranges from secondary to tertiary (via a 1,2-hydride shift). |
 |
| Qu20: |
We should work forwards, we are doing dehydrohalogenation of alkyl halides, i.e. an elimination of an alkyl halide to an alkene using a strong base / heat, an E2
elimination. We should note that it is then an anti-Zaitsev elimination giving the less highly substituted alkene. |
 |
CONFORMATIONAL ANALYSIS:
Understanding the terminology and the
energies involved in
conformational
analysis.
Qu 21:
The two Cl atoms are at 120 degrees (torsional angle) in this eclipsed conformation.
Qu 22:
The two bold bonds are at 180 degrees to each other and hence are anti in this chair conformation of cyclohexane.
Qu 23:
A careful comparison of the two structures shows that they are stereoisomers where the configurations at the alcohol bearing C are the same while the configurations at the fluoride bearing C are different. This makes them diastereomers.
Qu24:
First draw out the named structure in a favoured chair conformation. Trans- means that the two bromine groups are on opposite faces of the cyclic system and being 1,3- this means one axial and one equatorial.
Qu25:
Interpreting Newman projections... draw out the named structure first and see that it is a C6 structure. If one checks the C count in the Newman projections, only two of them are also C6. Then check the connectivity of those two structures.
Qu26:
The C atom with the Br attached is a chirality center, so use the Cahn-Ingold-prelog rules based on the heavier atom at the first point of difference to establish group priorities and the R or S configuration at the chirality center. Since the alkene has two methyl groups attached at the bottom alkene C, the alkene is not a stereocenter and therefore it does not require E or Z.
Qu27:
Isomers have the same molecular formula (C4H11NO), so simply check the molecular formulae...
Qu28:
Enantiomers are a type of stereoisomer, and require that we have the same overall connectivity other than 3D space, and we have the opposite configuration at the chirality center.
SPECTROSCOPY:
Use any IR information to get the
functional groups. Use the H-NMR
to get the number of types of H, how many of each type from the
integral
and what they are next to from the coupling patterns. Chemical shifts
should
tell you if the group is near -O- or maybe C=O groups etc.
| Qu29: |
IR shows a carbonyl (i.e. C=O) at 1735 cm-1 which is little high for a ketone. The C=O in the C-NMR peak is at 165 ppm suggesting a carboxylic acid or derivative. The C-NMR shows only 3 C types one being the C=O (165 ppm) and the other likely C=C (133 ppm). The H-NMR has just 2 types of H. The 1H singlet at 6.9 ppm,
suggests a -CH group maybe on C=C and the 3H singlet at 3.8 ppm,
suggests a CH3O-. Remember that one does not observe coupling between H of the same type. |
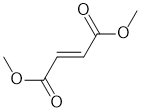
|
| Qu30: |
IR shows a carbonyl (i.e. C=O) at 1745 cm-1 which is high for a ketone. The C=O in the C-NMR peak is at 171 ppm suggesting a carboxylic acid or derivative. The C-NMR shows 5 C types including the C=O at 171 and 4 sp3 C 70-10 ppm. The H-NMR has 4 types of H. The 2H triplet at 4.1 ppm,
is consistent with a -CH2 group attached to something that deshields such as an O, the 3H triplet at 1.0 ppm,
is consistent with a -CH3 group connected to a -CH2 group. The 3H singlet at 2.0 ppm is consistent with a -CH3 group a C=O. |
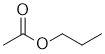 |
| Qu31: |
IR has no carbonyl (i.e. C=O) near 1715 cm-1 but shows a possible C-O at 1120 cm-1, no other groups are indicated. The C-NMR shows only 3 C types, all likely sp3, with one being deshielded (73 ppm). The H-NMR has 3 types of H. The 2H triplet at 3.4 ppm,
is consistent with a -CH2 group attached to something that deshields such as an O, the 2H sextet at 1.5 ppm,
is consistent with a -CH2 group next to 5H and the 3H triplet at 1.0 ppm is consistent with a -CH3 group next to a -CH2 group
which altogether means a -O-CH2-CH2-CH3. |
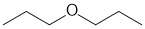 |
| Qu32: |
IR shows a carbonyl (i.e. C=O) at 1725 cm-1 which is a little high for a ketone. There are also IR absorptions at 1675 and 1645 cm-1 which could be another C=O and / or C=C.
The C-NMR shows 6 C types including peaks 198 ppm (suggests C=O in an aldehyde or ketone), 166 (possible C=O in a carboxylic acid or derivative), 140 and 131 (possible C=C). The H-NMR has 4 types of H. The 3H singlet at 2.3 ppm,
suggests an isolated -CH3 group, the 3H singlet at 3.8 ppm suggests a deshielded, isolated -CH3 group, and the 1H doublets at 6.9 and 7.3 ppm an unsymmetrical disubstituted alkene (H-C=C-H). These pieces of data are consistent with the keto-ester substituted alkene. |
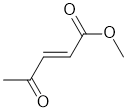 |
| Qu33: |
IR has no carbonyl (i.e. C=O) near 1715 cm-1 but it does have an absorption at broad 3400 cm-1 that suggests -OH or -NH. The C-NMR shows 3 C types. The H-NMR has 4 types of H. The 3H triplet at 0.9 ppm,
is consistent with a -CH3 group attached to a -CH2-, the 2H sextet at 1.6 ppm,
is consistent with a -CH2- group next to 5H and the 2H triplet at 3.6 ppm is consistent with a deshielded -CH2- group next to a -CH2- group
which altogether means a -CH2-CH2-CH3. The 1H broad singlet that is exchangeable supports it being an -OH (not an -NH2) |
 |
| Qu34: |
IR has no carbonyl (i.e. C=O) near 1715 cm-1 but it does have absorptions at 3370 & 3291 cm-1 that suggests an -NH2 due to the double band from the symmetric and asymmetric stretches. The C-NMR shows 3 C types. The H-NMR has 4 types of H. The 3H triplet at 0.92 ppm,
is consistent with a -CH3 group attached to a -CH2-, the 2H sextet at 1.45 ppm,
is consistent with a -CH2- group next to 5H and the 3H triplet at 2.65 ppm is consistent with a slightly deshielded -CH2- group next to a -CH2- group
which altogether means a -CH2-CH2-CH3. The 2H broad singlet that is exchangeable supports it being an -NH2 (not an -OH) |
 |