
Qu1: E
i has 3 types, ii has 2 types and iii has 5 types so iii
> i > ii
Qu2: B
Relates to hybridisation.
i is an aromatic CH with C = sp2, therefore 33% s character,
while ii and iii are both alkane CH, C = sp3, therefore
25% s character. Increasing the s character in the hybrid orbital makes the
orbital smaller and creates a stronger interaction with the H atom and so a
stronger bond so i is the strongest CH bond. Now how do we separate ii
and iii? ii is a secondary CH bonds whereas iii is a primary
CH bond. Primary CH bonds are stronger (from alkane stability or radical laboratory
expt). Therefore i > iii > ii
Qu3: AB
Stability of alkanes in terms of heats of combustion. More branched alkanes
are more stable so iii is the most stable and i is the least stable.
The more stable the alkane the less exothermic the heat of combustion. Therefore
iii > ii > i
Qu4: A
Calculate the formal charges for the atoms = group number - number of bonds
- lone pairs electrons or by relating to known structures. i = +1 based
on 6 - 3 - 2, ii = -1 (since treat Li as ionic) and iii = -2 based
on 6 - 0 - 8. Therefore i > ii > iii
Qu5: AB
The stronger the interaction of the orbital the stronger the bond and the lower
the energy of the orbital. The CH s bond is the strongest,
then the CC p bond. The CC p*
is a high energy antibonding orbital, so iii > ii > i
Qu6: A
Remember the rules for ranking resonance structures : complete octets are most
important. Here we focus on the ONO unit. i has a complete octets at
both O atoms and the N. ii has an incomplete octet on N and has one less
bond than i. iii is impossible because it has 5 bonds to N (i.e.
violates the octet rule). So i > ii > iii
Qu7: E
Draw each of them out in their lowest energy conformations. i
has an equatorial t-butyl group and an axial methyl group. ii has both
groups equatorial and iii has 3 equatorial and 2 axial methyl groups....
therefore there is a large 1,3-diaxial interaction destabilising iii.
Since substitutents generally prefer to be equatorial, ii is more stable
than i : hence iii > i > ii.

Qu8: AB
Basicity...either
think about the availability of the electrons in the base or the stability of
this base. i has a -ve O whereas ii has a -ve N and iii
a -ve C (sp3). These elements are all in the same row of the periodic
table, so electronegativity is a reliable factor. Since in terms of electronegativity
O > N > C and the more electronegative the atom the less willing it is
to share electrons, then in terms of basicity, C > N > O and so iii
> ii > i.
Qu9: E
Acidity....the
strongest acid will create the most of the conjugate acid. Compare to O systems
first... carboxylic acids, pKa = 5, are stronger acids than alcohols, pKa =
15, because of the resonance stabilisation in the carboxylate ion, so iii
> i. Now what about the alkyne? Alkynes
are not strong acids, pKa = 25 since the H is on a C and results in a carbanion
conjugate base. Therefore overall iii > i > ii
Qu10: E
Oxidation
states...count the bonds attached to each of the atoms to be considered.
A bond to a more electronegative atoms counts -1, a bond to the same type of
atom counts 0 and a bond to a less electronegative atom (e.g. H) counts +1.
Total the count and then since the atoms are all neutral, just switch the sign
since the oxidation state for the central atom plus the groups attached must
equal 0. In i the C is attached to 2 O (count - 2) and 2 H (count +2)
therefore oxidation state C = 0. In ii the O is attached to C(count +1)
and 1 bond to O (count 0) therefore oxidation state O = -1. In iii the
C has two bonds to C (count 0 per bond), and a bond to Cl (count -1) therefore
oxidation state C = +1. Therefore iii > i > ii
Qu11: AB
5% NaOH (aq. base) will react with carboxylic acids and phenols to form
their ionic conjugate bases which will be soluble in the polar aqueous medium.
Hence A and B dissolve.
Qu12: C
Melting points are not sensitive to atmospheric pressure (it's boiling
points that need to be corrected).
Qu13: A
A % yield calculation. Reaction stoichiometry is 1:1. Toluene has MW = 92.141
g/mol so 0.92g = 0.01 mol. Bromine has MW = 159.808 g/mol so 1.0g = 0.0063 mol.
(bromomethyl)benzene has MW = 171.037 g/mol so 1.02g = 0.0059 mol. Hence bromine
is limiting and the yield is 0.0059 / 0.0063 = 95%
Qu14: D
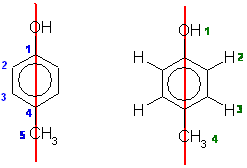
From the molecular models experiment. First draw out 4-methylphenol (if
you can't, then you need to work on your nomenclature).
The number of types of C are shown below in blue on the left and the H are shown
below in green on the right. The red line shows the symmetry due to the mirror
plane that bisects the molecule.
Qu15: B or E
If the crystals are already present then all that is needed is filtration,
but otherwise recrystallise.
Qu16: BE
A is false because the
Rf is given by the distance traveled by the sample / distance graveled by the
solvent from the origin. C is false because two samples could have the
same Rf even if they are not identical. D is false because two spots
suggests two materials which means it would be impure.
Qu17: A
Oxidation
states
N7 has the following bonds : 2 x C (count +2), and 1 x H (count +1) so
the sum = +3 therefore the oxidation state of N7 = -3.
N12 has the following bonds : 4 x C (count +4) so the sum = +4 but the
charge = +1 therefore the oxidation state of N12 = -3
Qu18: A
The functional
group in question is O=C-N so it's an amide.
Qu19: D
The amine
has 4 alkyl groups attached so it's a quaternary amine.
Qu20: E
IHD....Unsaturation
arises due to pi bonds or rings. So count these up in the structure:
there are 9 C=C, 2 C=O plus 3 rings, so 14 in total.
Qu21: B
N7 is part of an amide so it is sp2 (due to the
resonance interaction), N12 is sp3 it's
an amine N and O10 is part of a double
bond so it is sp2.
Qu22: E
Basicity...look
at the stability (electron availability) at each site. N7 is part of
an amide, so the N lone pair is less available because it's involved in resonance.
O10 is a carbonyl, two lone pairs in sp2 orbitals. C11 doesn't
have lone pairs - the electrons are all involved in CH bonds. N12 does
have a lone pair - the electrons are all involved in NC bonds and then O37
has three lone pairs and a -ve charge... this is where the H+ would add.
Qu23: A
Hybridisation.
C1 has 4 groups attached (3 x H, 1 x C) = sp3, C2 has
3 groups attached (2 x C, 1 x O) = sp2 and C4 has 4 groups attached (1
x H, 2 x C and 1 x lone pair) - but the lone pair is involved in resonance with
the adjacent pi system of the carbonyl = sp2.
Qu24: A
Oxidation
states. C1 has bonds to 3 x H + 1 x C = +3 + 0 therefore = -3, C2
has bonds to 2 x C + 2 x O = 0 + -2 therefore = +2 and C4 has bonds to
1 x H + 2 x C = +1 + 0, but it has a charge of -1 therefore its oxidation state
= -2 (to allow for the -ve charge).
Qu25: E
Note that this is an eclipsed
conformation - the two C-Cl bonds have a 120o torsional
angle.
Qu26: D
We need to recgonised that this is a Newman
projection of a tetrahedral system so the bond angle will be the normal
tetrahedral angle of 109.5o
Qu27: D
Trans-1,3-dimethylcyclohexane has one eq. and one axial group. A
is trans-1,4-, B, C and E are cis-1,3-.
Qu28: D
If you look at the Newman projection or build a model and look at it, the
axial CC bond is at f 60o and hence it is gauche. Staggered,
while describing the general conformation is not the most specific term therefore
it is not the best term.
Qu31: D
The boat
(C, D) conformations are less stable than the chairs
(A, B, E). Putting the methyl group "inside"
the boat increases the flagpole interaction hence D is the least stable.
Qu32: A
All are trimethyl substituted cyclohexanes.... since the methyl group prefers
to be equatorial, the system with the most equatorial methyl groups will
be the most stable (lowest energy).
Qu33: E
Longest chain is C7, contains a C-C only so we have a heptane with 2 methyl
groups and an ethyl group. A is
wrong because the longest chain (pent = 5) is wrong. B and C are wrong because the longest
chain (hex = 6) is incorrect. D is wrong because the alphabetisation
is incorrect : ethyl is before dimethyl.
Qu34: C
Longest chain is C9, including a CC triple bond so we have a nonane with a methyl
group and an ethyl group. Numbering dictated by the alkyne to be as low
as possible : 4-yne. A is wrong because alkynes are not named E
or Z as they are linear, B is wrong because the numbering is wrong.
D is wrong because the longest chain is not
C2. E is wrong because the longest chain is not
C8 (oct).
Qu35: A
The compound is an acyl halide, -C(=O)-Cl - the carbonyl group makes a
difference and the carbon of the carbonyl is C1. It is not a ketone, an
aldehyde or an ether.
Qu36: E
Alkene stereochemistry as described by E and Z. The longest chain including
the C=C and the C=O is 5 carbons so we need a pentene. The functional group
is an aldehyde so need the suffix -al, this rules out A and C.
The double bond has E stereochemistry because the higher priority groups (the
CHO and the Cl) are on opposite sides of the double bond. The aldehyde C defines
C1.
Qu37: E
N-benzyl....amide implies that we have a C6H5CH2- group attached to a nitrogen
in an amide. A is a nitrile. B and C are N-ethyl systems. D is N-phenyl
NOT N-benzyl.
Qu38: D
Use the descriptors cis- and substituent positions (1,2) and look at
the position of the two alkyl groups.... A trans-(1,2)-, B
trans-(1,3)-, C trans-(1,2)- and E is cis-(1,3).
Qu39: B
The name is a pentanal so we need a C5 chain including the C=O and one hydroxy
group. The aldehyde C is C1, so this defines the -OH group location. C
is wrong because it is a ketone and E
is wrong because it is has a 4-hydroxy. In terms of assigning
configurations, the group order is -OH > CH2CHO > CH2CH3 > H.
Remember to assign the sense of the rotation to the order of the groups when
the H is away from you...A, D and E are all R.
Qu40: A
Did you look at the nomenclature
of polycyclics? This is a bridged
system. The [3.1.0] means that there are 3C and 1C in the links between
the common C atoms. This gets rid of E which is [3.2.0]. Note that the
hexane means we have 6 C in the parent ring structure and the bicyclo means
two rings. C is a tricyclo. Then we number from a bridgehead C
bearing in mind the first point of difference rule.
B is 3,6-dimethylbicyclo[3.1.0]hexane and D
is 1,3-dimethylbicyclo[3.1.0]hexane.
![[Chem 350 Home]](mol.gif)