Here is an post-mortem analysis / "how to" for the MT. The questions are split by the sections. At the start of each section are a few suggestions of what to look for or how to tackle the question type.
Qu1: D
Question about geometry goes back to hybridisation and geometries of
cycloalkanes i is a cycloalkane with bond angles of 60o,
ii
is an alkyne so the central C is sp , i.e. linear, with bond
angles
of 180o, and iii is an alkene so the central C
is sp2, i.e. trigonal planar, with bond angles of 120o,
therefore giving : ii > iii >
i
Qu2: A
Just have to draw them out, no quick way. Constitutional isomers
means different numbers and types of bonds giving different functional
groups and branching patterns. C4H10O is
saturated,
it could be 1-butanol, 2-butanol, 2-methyl-1-propanol,
2-methyl-2-propanol,
diethyl ether, methyl n-propyl ether, isopropyl methyl ether (7
isomers).
C5H12 could be pentane, 2-methylbutane,
2,2-dimethylpropane
(3 isomers). C3H6 is unsaturated it could be
propene
or cyclopropane (2 isomers), therefore i > ii > iii
Qu3: C
First you need to complete the formal charges, then apply the rules
for ranking resonance structures. In these three, the C, N and O
all have complete octets in all three structures. ii is
the major contributor since it has no formal charges. In i
the O is negative and N positive which matches the relative
electronegativities
whereas in iii the O is positive and N negative which opposes
the
relative electronegativities, so i is more important than iii.
So overall we have ii > i > iii
Qu4: C
Calculate the formal charges for the C atom = group number - number
of bonds - lone pairs electrons : i = neutral based on 4 - 3 -
1,
ii
= +1 based on 4 - 3 and iii = -1 based on 4 - 3 - 2.
Therefore
ii
> i > iii
Qu5: AB
Acidity .... consider the general acidity equation HA <=>
H+
A-. Look at the factors that stabilise the conjugate base,
A-. All C-H bonds, but the hybridisation of the C atom changes as
we go alkane to alkene to alkyne. Think about how the lone pair
of
electrons that results in the conjugate base is stabilised by being in
sp3 vs sp2 vs sp hybrid orbital. The
critical
factor is the % s character (25%, 33%, 50% respectively). More s
character
means a less diffuse orbital and puts the electrons closer to the
positive
nucleus and hence means there is more stabilisation. The greater the
stabilisation
of the conjugate base. So we have iii > ii > i
Qu6: AB
The easiest way to count up the valence electrons here is to use the
molecular formulae of each compound and use the valence electron count
for each element (H = 1, C = 4, N = 5, O = 6, F = 7) then
remember
to adjust the overall count to match the systems overall charge. This
way
you get i = 23 +1 = 24, ii = 26 -1 = 25, and iii
=
26. Therefore iii > ii > i
Qu7: B
Cycloalkane stability so need to think of ring strains.
Cyclohexane
is the most stable system (lowest energy) so its DHfwill
be the most exothermic. Cyclopropane is highly strained and is the
least
stable, so its DHfwill
be the least exothermic. This means we have i > iii
>
ii
Qu8: A
Boiling point is controlled by intermolecular forces. For non-polar
alkanes this is the weak induced dipole interactions. More branched
structures
tend to be more spherical in shape and hence have less surface areas in
contact with other molecules and so less energy is required to separate
them. Hence they have lower boiling points. (Remember physical
properties
are not connected to thermodynamic stability which is controlled by the
forces that hold the atoms in the molecules together). Therefore
here we have i > ii > iii
Qu9: A
Either use the equation that relates energy of a photon to frequency
E = h n and c = l n
or just use the fact the frequency, n, is directly
proportional to E, or use the fact that i corresponds to
visible
light, ii is a "classic" IR frequency for a C=O stretch and 60
MHz
is the frequency of a low end H NMR in the radio frequency region.
Converting
to frequencies, 500 nm = 6 x 1014 Hz, 1715 cm-1
corresponds
to a wavelength = 5831 nm = 5.15 x 1013 Hz and 60 MHz = 60
x
106 Hz. Any of the methods should lead you to the
energies
being i > ii > iii
Qu10: D
Look at the relative positions of the two alkyl group substituents.
In i it is staggered and they are anti (180o) , in ii
it is eclipsed and they are syn (0o) and in iii
it is staggered and the groups are gauche (60o). Staggered
conformations
are more stable than eclipsed and strain is lowest when the biggest
substituents
are well separated. Hence ii > iii > i
Qu11: B
From the extraction experiment. Seems a hard question needing math,
but if you understand the principles, it's pretty straight forward.
Given
that KD = 1 and equal volumes of solvents are used it means
the relative amounts in the two layers are the same, so the amount
extracted
each time is half. Do two extractions means you extract 50% then 50% of
50% i.e. 25%, so the total amount extracted is 75%. See,
that
wasn't too difficult was it ?
Qu12: B
From the physical properties experiment. Remember boiling points
increase
with increasing pressure so they are higher at sea level than here in
Calgary
at almost 4000ft above sea level. Rough rule of thumb for Calgary is 1o
for every 15o above 50o. Thus for 200o
we are talking about a 10o increase.
Qu13: BC
The distillation experiment. Slow patient heating ensures a good
thermal
equilibrium is set up in the fractionating column to facilitate the
separation
process. A longer column means there are more evaporate -
condense
"cycles" or theoretical plates meaning improved separation. There
is no cooling water in the fractionating column and cooling the column
would not allow any separation as the vapours would reflux rather than
reach the top of the fractionating column and enter the condenser.
Qu14: E
Simple or vacuum filtration may not remove the impurity, it would need
to be soluble while the desired compound would need to be insoluble.
Distillations
are most commonly used for miscible liquids. Recrystallisation would
mean
that the impurity would remain in solution. Evaporation would not get
rid
of a non-volatile impurity.
Qu15: E
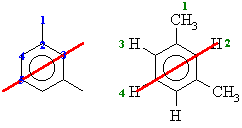
From the molecular models experiment. 1,3-dimethylbenzene the
number of types of C are shown below in blue on the left and the H are
shown below in green on the right. The red line shows the symmetry due
to the mirror plane that bisects the molecule.
Qu17: C
C13 is attached to 1 x O = -1, 1 x H = +1 and 2 x C = 0 so the
sum = 0, therefore C13 = 0
Qu18: CDE
First fill in the lone pairs on the heteroatoms one on each N and two
on each O. N1 has 2 attached groups and a lone pair = sp2,
C5
has 3 attached groups = sp2, O14 has 2 attached
groups
and two lone pairs = sp3, N16 has 3 attached groups
and
a lone pair = sp3, and C21 has 4 attached groups
(don't
forget the 2H atoms) = sp3.
Qu19: CD
A tertiary C will have 3 other C atoms attached to it. C2
has only 1 C attached, C12 has no C attached, C19
has 3 C attached, C20 has 3 C attached and C22
has 2 C attached.
Qu20: BD
Ortho positions are those adjacent to the substituent.
Qu21: AD
Alcohol, ROH at O14, no amides RCONH2 or esters RCO2R.
Ethers, ROR at C7-O11-C12, and no phenol, ArOH.
Qu22: D
Count the rings and the p bonds to get 4
rings plus 6 p bonds = 10
Qu23: E
Since all are CC bonds we need to consider the hybridisation of the
atoms involved and the type of bond. A double bond will be shorter than
a single bond due to the increased bonding interaction. C8-C9
is sp2 to sp2 (benzene so between C-C and C=C in
character), C4-C13 is a single bond sp2 to sp3,
C13-C15
is a single bond sp3 to sp3,
C20-C23
is a single bond sp3 to sp2,
C23-C24
is a double bond sp2 to sp2. Therefore C23-C24
will be the shortest.
Qu24: D
All the other options are examples of stereoisomers that have the same
groups and bonds and differ only on how they are connected spatially.
Qu25: C
A and D are both cis. B is trans-1,2-.
So C or E ? Put the larger tBu group in the more
favourable
equatorial position rather than the axial position.
Qu26: A or B
Credit was given for either A or B as with hindsight,
the question is potentially ambiguous depending on whether you decide
to
look at the worst single 1,3-diaxial interaction (B) or
worst
total 1,3-diaxial interactions (A). Larger substituents have a
strong
perference to be equatorial because of unfavoutable Van der Waals
strain
with the other axial groups when they are axial themselves.
Qu27: A
It shows a staggered conformation where the two methyl groups are at
180o, so anti is the best term to use here. B syn
means
the two methyl groups are at 0o, C gauche means the
two
methyl groups are at 60o, D eclipsed is the
more
general description when the three bonds off the front carbon are
aligned
with the three off the rear carbon, E cis and AB
trans
are used to describe a pair of substituents on a C=C or in a cyclic
system.
Qu28: C
See the images in the etext
Qu29: C
There are 10 C atoms in the decalin
system, each with one axial and
one equatorial substitutent. The two F atoms in this molecule are both
axial and there are no other axial substituents other than H atoms. So
10 - 2 = 8.
Qu30: E
Longest chain is C6, contains a C=C so a hexene. If alkene is the main
group, then its a 2-ene. Dimethyl substituted amino group hence
N,N-dimethylamino,
located at C4. Given that the C substituents on the C=C out rank
the H atoms, the higher priority groups are on opposite sides of the
alkene
so it is an E configuration.
Qu31: C
Alcohol group at C1, then use first point of difference to get the
methyl group at C3, ethyl at C4. Remember to alphabetise.
Qu32: B
An ester not an ether, so it can't be A or C. The
carbonyl
defines the acid "half" of an ester, so here, where it is attached to
the
benzene ring, we are looking at a benzoate ester. In this case
the
straight chain in the other half, the alcohol "half", means we have the
n-butyl ester. Thus overall we have n-butyl benzoate.
Qu33: A
The alkene has an E configuration (C > H and CºC
> CH3). The longest chain is C6, so a hexenyne (note
order alkene
- alkyne). Since the alkyne is terminal it is numbered as "1" so we
have
a hex-3-en-1-yne... now just locate the two methyl groups at C3 and C5.
Qu34: B
Complex substituents in an alkane... locate longest chain, here C11,
identify substituents, number based on first point of difference (i.e.
5,6- rather than just 6,7- diethyl groups). The complex substituent has
a C2 chain (so ethyl) with two methyl group at C1 so 1,1-dimethylethyl
located at C8 on the main chain.... Remember to alphabetise....prefixes
such as "di-" don't count normally, but they do if they are part of the
bracketed term as in complex substitutents, hence "dimethyl" preceeds
"ethyl"
and then "methyl" so B is correct and D has an error in
the
alphabetisation. We could consider naming as a tert-butyl (e.g. as per
C) but this name is incorrect based on the first point of
difference
rule for the locants (it would have needed to be
8-tert-butyl-5,6-diethyl-4-methylundecane).
Qu35: D
Meta implies we have 1,3-substituents so that restricts us to
C,
D
or E. A and C are amines, B and D
ares
amides and E has a nitro group.
Qu36: B
Use the descriptors trans- and (1,3) and look at the position
of the two methyl groups.... A cis-(1,3)-, B trans-(1,3)-,
C
trans-(1,2)-,
D
trans-(1,4)-, and E is
trans-(1,4).
Qu37: B
C and D are not carboxylic acids. The others are
stereoisomers.
The relative priority of the groups (Cahn-Ingold-Prelog rules) at the
chirality
center (4 different groups attached) is : OH > C=C > CH2
> H.
A and E where the sense of rotation of the three higher
priority
groups is counter clockwise are therefore both S. B is R
(remember
to put the lost priority group, the H, at the back when determining the
sense of rotation).
Qu38: A
Did you look at the nomenclature of bicyclics ? Bicyclic means two
fused rings. The [3.2.1] means that there are 3C, 2C and 1C in the
links
between the shared (bridgehead) C atoms. This gets rid of C, D,
and E which are [2.2.1], [2.2.2] and [2.2.2] respectively. We
number
from one of the shared C atoms via the longest link first so as to give
the alkene the lowest number.
![[Chem 351 Home]](../mol.gif)